Corona_Fakten: 2. fáze kontrolního experimentu – Vyvráceno: Jak byla autoritativní studie o SARS-CoV-2 zfalšována čínskými vědci

3. dubna 2022
Přeložil: Vladimír Bartoš

Když vědci ohýbají data na maximum a ostatní je následují
Před několika dny jsme na našem kanálu Corona_Fakten zveřejnili náš kontrolní experiment fáze 1 (CZ verze ZDE), ve kterém jsme diskutovali o laboratorních výsledcích takzvaného cytopatickém efektu.
Získané výsledky jasně ukazují, že tento efekt nelze považovat jako důkaz přítomnosti viru – jak tvrdí virologové od roku 1954 – ale že k efektu vede samotné uspořádání experimentu, i když není přidán žádný “infikovaný materiál”.
Ti, kteří ještě nejsou obeznámeni s těmito kontrolními experimenty, je mohou podrobně prostudovat v [14].
Dnes vám představujeme pokračování: kontrolní experiment fáze 2.
Je zcela běžné, že jedna studie slouží jako základ, na jehož zjištění pak provádějí výzkum všichni ostatní vědci a SARS-CoV-2 není jiný.
Není však těžké si představit dalekosáhlé důsledky, pokud právě tato studie, která v tomto případě diktuje všem ostatním na celém světě, jak by měl údajný virus způsobující onemocnění geneticky vypadat, vytvořila falešné a chybné údaje pomocí nevědeckých metod.
Mělo by být zřejmé, že se automaticky spustí obrovská lavina chybných následných vědeckých studií. S dobře známými důsledky, jichž jsme byli svědky v posledních dvou letech v přímém přenosu.
Zatímco v případě viru spalniček to byla studie Johna F. Enderse z roku 1954, která se stala “matkou” všech dalších studií, v případě SARS-CoV-2 hraje hlavní roli vědecká studie profesora Zhanga a kol, která byla publikována v časopise Nature a nese následující název:
“Nový koronavirus spojený s lidským respiračním onemocněním v Číně” (“A new coronavirus associated with human respiratory disease in China”) [1].
Nyní jsme studii profesora Zhanga a kol. rozpitvali do nejmenších detailů a museli jsme si šokovaně přiznat, že čínští vědci nejenže nechali projít značné nedostatky, ale oni dokonce nebyli tak dobří, aby “podváděli”! Co jiného to je, když to není podvod?
- Polovina všech publikovaných sekvencí byla začerněna, takže původní nukleotidovou sekvenci již nelze získat, což znemožňuje vědcům rekonstruovat sekvenci. [13]
- Žádný virus nebyl izolován, ani odstředěn, natož sedimentován.
- Sestavení, tj. konstrukci genomu SARS-CoV-2, který se stal vzorem pro všechny vědce na celém světě, nelze reprodukovat pomocí zveřejněných čínských sekvencí.
- Neexistuje žádný důkaz, že by tvrzený genom SARS-CoV-2 byl vytvořen z čisté virové RNA.
- Rafinovaný vliv čínských autorů spočíval v tom, že se vzhledem ke znalosti anamnézy a symptomatologie posuzovaného pacienta soustředili pouze na hledání možných respiračních patogenů.
- Číňané dosáhli dvou zcela odlišných konečných výsledků. Nejdelší souvislá sekvence nalezená překrytím pomocí programu “Megahit” byla 30 474 nukleotidů, zatímco druhý program “Trinitiy” vytvořil ze stejného souboru dat nejdelší kontig o délce 11 760 nukleotidů. Naopak Trintiy vytvořil výrazně více souvislých sekvencí, konkrétně 1 329 960 kusů, než Megahit (384 096). To je obzvláště důležité z vědeckého hlediska, zejména z hlediska reprodukovatelnosti.
- Čínská sekvenační data zpřístupněná veřejnosti nejsou původní, nezpracovaná sekvenační data.
- Příčinná souvislost tohoto onemocnění nebyla popsána, protože se ji autorům nepodařilo zjistit. Uvedené zjištění tedy představuje pouze velmi slabou korelaci, protože byl vyšetřen pouze JEDEN pacient!
- Sekvence pravděpodobně obsahují i sekvence lidského původu, přesněji: ribozomální RNA (pokud jsou virologické databáze správné). Při depleci RNA tedy nebyla odstraněna veškerá ribozomální RNA.
- Pokrytí cílového genomu spojenými krátkými sekvencemi (PCR primery) vykazuje velmi nerovnoměrné rozložení. Při nestranném sekvenování by se dalo očekávat, že přibližně každý nukleotid bude pokryt podobným počtem nalezených sekvencí. Za zjednodušeného předpokladu binomického rozdělení by se dalo očekávat, že pokrytí bude ležet převážně v zobrazeném koridoru. Výrazné rozdíly v pokrytí vyvolávají otázku, zda některé sekvence, které nebyly zahrnuty do původního vzorku, nebyly náhodně vytvořeny během sekvenování.
- Nebyly provedeny žádné kontrolní pokusy.
Některé příklady nezbytných kontrolních pokusů, které virologové neprovedli:
- Kontrolní experimenty, které vylučují možnost, že cytopatický účinek, o němž virologové tvrdí, že je důkazem viru, není způsoben samotným experimentálním designem. V některých studiích se tvrdí, že byla provedena kontrola, která však nebyla nikdy zdokumentována, což ji činí nedostupnou pro kontrolu.
- Druhým kontrolním pokusem, který vyplývá z vědecké logiky, je intenzivní použití vyvinutého postupu PCR (real-time RT-PCR) s klinickými vzorky od lidí s jinými chorobami, než které jsou přisuzovány viru a se vzorky od zdravých lidí, zvířat a rostlin, aby se ověřilo, že jejich vzorky nejsou také “pozitivní”.
- Proveďte kontrolní experimenty, abyste vyloučili možnost
- že jde o lidskou/mikrobiální RNA z výplachu plic zdravé osoby,
- osoby s jiným plicním onemocněním,
- osoby, která byla negativně testována na SARS-CoV-2..,
- nebo z takové RNA z rezervních vzorků odebraných v době, kdy virus SARS-CoV-2 ještě nebyl znám,
- je možné přesně stejné sestavení genomu viru z krátkých fragmentů RNA!
- Kontrolní experimenty týkající se sekvenování genomu z “infikované buněčné kultury”, aby se vyloučilo, že lze sestavit i jiné virové genomy “de novo” nebo zarovnáním s jinými referenčními genomy.
- Kontrolní experimenty k vyloučení, že genom cílového viru byl sestaven “de novo” nebo zarovnáním z negativní kontrolní kultury.
V tuto chvíli se vám jako opravdovému vědci skutečně zatají dech, protože to odhalilo další potvrzení toho, že všechna pravidla vědy byla porušena a nerespektována.
Může mi někdo vysvětlit, proč si žádný vědec, který vycházel z práce Číňanů, nevšiml obrovských nedostatků? Nebo je prostě s nebývalou drzostí ignorovali? Nebo existovaly pobídky, aby se dívali jinam?
Podle hesla: hlavní je, že si připisuji zásluhy, protože jsem první!
Nový koronavirus spojený s lidským respiračním onemocněním v Číně – Byl ve Wuhanu nalezen “nový” a “choroboplodný” virus?
Část A: Jak byla určena sekvence “nového” koronaviru SARS-CoV-2?
Část B: Kritický přehled metod a závěrů
Úvod
V současné pandemii zaujímají významnou pozici lékařští vědci, především virologové. Jejich hodnocení se v naší společnosti téměř nezpochybňuje a rozhodujícím způsobem se jím zdůvodňují přijatá opatření. Tento přístup se nezdá být adekvátní, zejména s ohledem na obrovský dopad na společenský život. Pokud jako společnost otevíráme významný prostor pro technokratické myšlení a jednání, které formuje společenský život, musíme mít také potřebné znalosti a odpovídající metodologické dovednosti ve všech oblastech, zejména v řadách rozhodovacích orgánů. Jinak víra předchází poznání. Jedná se o problém, který se v současné době dramaticky odhaluje a jeví se jako vysoce toxický. Nyní je čas, aby mnoho lidí dospělo, vydalo se na výzkumnou cestu a uvěřilo svým vlastním schopnostem. Nejde o nedůvěru, ale o zpochybňování. Absolutní pravdy, o níž není pochyb, nemůžeme dosáhnout ani ji očekávat, pokud někde existuje. Poznatky je třeba neustále zpochybňovat, testovat a odhalovat dogmata. V těchto důležitých atributech spočívá motor vědecké práce. Měli bychom být vždy otevření a vděční za nové myšlenky, argumenty a interpretace domněle známého, protože ty obohacují náš život jako povzbuzující elixír.
Pochybovat o známém může být odvážnější než zkoumat neznámé.
Alexander v. Humboldt
Tento článek se podrobně zabývá vědeckou publikací “A new coronavirus associated with human respiratory disease in China” [1], ve které byla poprvé navržena genetická sekvence údajného “nového” koronaviru SARS-CoV-2. Tato práce byla publikována 3. února 2020 ve vědeckém časopise Nature.
Je rozdělený na dvě části. V první části je vysvětlen metodický postup konstrukce tvrzeného virového genomu SARS-CoV-2. V druhé části následuje kritická analýza publikace z vědeckého hlediska. Poukazuje se tak na metodologické nedostatky, kritické otázky a možnosti kontroly, které srozumitelným způsobem zpochybňují objev “nového” a “choroboplodného” viru.
Část A Jak byla určena sekvence “nového” koronaviru SARS-CoV-2?
Výchozí situace
Byl vyšetřený 41letý pacient, který byl 26. prosince 2019 přijat do Ústřední nemocnice Wuhan. Byl zaměstnancem místního rybího trhu, který je podle epidemiologického šetření původním místem výskytu “nového” viru. Pacient byl přijat s teplotou nad 37,5 °C, tísní na hrudi, kašlem, bolestí a celkovou slabostí. Předběžná etiologická vyšetření vyloučila mimo jiné přítomnost chřipkových virů. Byly vyloučeny i další běžné respirační patogeny, jako jsou lidské adenoviry. Poté, co se pacientův stav ani po podání antibiotik a antivirotik nezlepšil, byl převezen na jednotku intenzivní péče a později do jiné nemocnice ve Wuhanu k další léčbě. Za účelem identifikace možných patogenů spojených s tímto onemocněním byla pacientovi odebrána tekutina z bronchoalveolární laváže (BALF), která byla následně metatranskriptomicky sekvenována.
Metody
Vědecké publikace v oblasti virologie obvykle obsahují oddíl o metodách, v němž je postup podrobněji popsán. Jsou zde popsány například metodické postupy a použité sekvenační technologie a také informace o bioinformatických protokolech.
Vykazování údajů
Jak již bylo uvedeno, byla vyšetřena přesně jedna (!) osoba. Provedené experimenty a vyhodnocení výsledků nebyly prováděny naslepo. Všichni zúčastnění vědci měli úplné informace, zejména znali klinický obraz pacienta.
Informace pro pacienty a odběr vzorků
Studie se týkala pacienta s akutním nástupem horečky (teplota nad 37,5 °C), kašlem a svíravým pocitem na hrudi, který se dostavil do Ústřední nemocnice ve Wuhanu v Číně. Při příjmu byla pacientovi odebrána bronchoalveolární lavážní tekutina (BALF), která byla uchovávána při teplotě -80 °C až do dalšího zpracování. Veškeré informace o pacientovi, jako jsou údaje o vyšetření a laboratorní údaje, byly k dispozici ve formě klinických záznamů.
Vzorek pacienta a sekvenování RNA
Nejprve byla z BALF extrahována celková RNA (ribonukleová kyselina). K tomuto účelu se používají komerční extrakční soupravy. Tyto soupravy se obvykle používají podle pokynů příslušného výrobce. Po extrakci bylo před následným sekvenováním posouzeno množství a kvalita roztoku RNA. Než však bude možné RNA sekvenovat, musí být vytvořena tzv. knihovna RNA. Zde je dostupná RNA technicky připravena pro krok sekvenování. RNA musí být například přepsána do cDNA pomocí reverzní transkripce a rozdělena na krátké fragmenty. V tomto případě byla ribozomální RNA odstraněna během konstrukce knihovny podle pokynů výrobce. Ribosomální ribonukleová kyselina (rRNA) se nachází v lidských ribozomech, kde probíhá biosyntéza bílkovin. Před vlastním sekvenováním se krátké fragmenty amplifikují pomocí polymerázové řetězové reakce (PCR), aby je použitý sekvenátor mohl detekovat a určit sekvenci nukleotidů. Sekvenování knihovny RNA bylo provedeno na platformě MiniSeq společnosti Illumina v Šanghaji.

Zdroj [2]
Bioinformatické zpracování přečtených sekvencí a identifikace virů.
Výsledkem sekvenování podle výše popsaného protokolu a následné úpravy a kontroly kvality pomocí programu Trimmomatic bylo celkem 56 565 928 krátkých sekvencí (čtení) o délce přibližně 150 nukleotidů. Jak bylo vysvětleno výše, krátké sekvence lze popsat pomocí sekvencí písmen, které se skládají ze čtyř nukleotidů A, T, C, G.
Sekvence písmen jsou uloženy v běžných textových souborech. V tomto případě byla krátká čtení uložena ve formátu FASTQ [3], který obsahuje další informace o kvalitě jednotlivých nukleotidů. Z těchto informací o kvalitě každého jednotlivého nukleotidu v rámci přečtené krátké sekvence lze určit pravděpodobnost, že příslušný nukleotid byl sekvenátorem správně zaregistrován.

Nyní se pokusíme určit delší souvislé nukleotidové sekvence z krátkých sekvenčních čtení (56 565 928 čtení x 150 nt dlouhých). Za tímto účelem se hledají překryvy mezi krátkými sekvencemi.
Vzhledem k velkému počtu krátkých čtení, v tomto případě asi 56 milionů, jsou pro tento krok zapotřebí bioinformatické programy a výkonné počítače.
Delší nukleotidové sekvence vypočtené překrýváním se nazývají kontigy. Slovo contig pochází z anglického slova contiguous, což znamená sousedící. Tento proces znázorňuje následující schéma.

Ve zde posuzované publikaci bylo komplexní generování kontigů provedeno pomocí dvou tzv. assemblerů Megahit a Trinity ve verzích v.1.1.3 a v.2.5.1.
Pro získání delších kontigů z milionů krátkých sekvenčních čtení bylo použito výchozí nastavení. Předtím však byly odstraněny možné sekvence lidského původu pomocí lidského referenčního genomu (lidská verze 32, GRCh38.p13) [4]. Po tomto kroku zůstalo 23 712 657 krátkých sekvencí, které nejsou lidské (dosud známé) (při dalších kontrolních experimentech bylo prokázáno, že byly lidského původu, viz ZDE, pozn. překl.). Následující tabulka uvádí přehled obou sestav.

Tabulka 1: Přehled výsledků generování kontigů pomocí Megahitu a Trinity.
Porovnání kontigů se známými sekvencemi v příslušných nukleotidových a proteinových databázích odhalilo vysokou podobnost s netopýřím virem podobným SARS SL-CoVZC45, MG772933, a to v případě nejdelších kontigů v každém případě (Megahit: 30 474 nt a Trinity 11 760 nt). Delší ze dvou kontigů o délce 30 474 nukleotidů podle autorů pokrýval téměř celý virový genom a byl použit pro návrh primerů pro tzv. následnou konfirmaci PCR a určení délky genomu.
Virový genom byl uspořádán pomocí zarovnání sekvencí dvou reprezentativních beta-koronavirů, a to SARS-CoV Tor2, AY274119 a netopýřího SL-CoVZC45, MG772933. První je spojen s lidmi a druhý s netopýry.
Dostupnost dat
Sekvenční čtení byla uložena v databázi NCBI Sequence Read Archive (SRA) pod přístupovým číslem BioProject PRJNA603194. Kompletní sekvence genomu WHCV (nyní SARS-CoV-2) byla uložena v GenBank pod identifikačním číslem MN908947.
Kontrolní zkoušky
V této publikaci “A new coronavirus associated with human respiratory disease in China” [1] nejsou doloženy žádné kontrolní pokusy.
Část B Kritický přehled metod a závěrů
Poté, co byl podrobně prozkoumán postup určení virového genomu údajného “nového” a “choroboplodného” viru SARS-CoV-2, je v této části uveden kritický přehled jednotlivých kroků. Zpochybňování vědeckých poznatků je elixírem živé vědy.
Poznámky: Vykazování dat
Zúčastnění vědci nebyli slepí a proto znali anamnézu a symptomatologii zkoumaného pacienta. To pravděpodobně ovlivnilo autory, aby hledali možné respirační patogeny. Již v tomto okamžiku by existovala jednoduchá možnost kontroly.

Poznámky: Vzorek pacienta a sekvenování RNA
Jak je podrobněji vysvětleno v části “Vzorek pacienta a sekvenování RNA”, před vlastním sekvenováním, tj. stanovením nukleotidové sekvence krátkých fragmentů, je nutné provést některé technické kroky.
Například komerční soupravy se používají – k extrakci celé RNA ze vzorku, – k odstranění ribozomální RNA ze vzorku, – k převodu RNA na cDNA, – k fragmentaci RNA nebo cDNA na krátké části nebo – k amplifikaci (PCR) cDNA, aby bylo možné určit nukleotidovou sekvenci.
K testování množství a kvality RNA se používá komerční technické vybavení. Jedná se tedy o složité procesy, které mohou vědci ovlivnit jen ve velmi omezené míře. To přímo vede k další možnosti kontroly, která není ve vědecké literatuře doložena.

Je tedy třeba poznamenat, že sekvenování RNA z dostupné bronchoalveolární laváže vyžaduje velmi složitý protokol. Prováděné kroky jsou obtížně kontrolovatelné. Je proto otázkou, do jaké míry lze zaručit reprodukovatelnost, která je nezbytná pro vědeckou kvalitu, a to bez znalosti referenčního genomu.
Poznámky: Bioinformatické zpracování přečtených sekvencí a identifikace virů
Především je zarážející zřetelný rozdíl mezi výsledky obou sestav Megahit a Trinitiy. Rozdíl mezi oběma výsledky je obrovský. Například nejdelší souvislá sekvence nalezená překrýváním s Megahitem byla 30 474 nukleotidů, zatímco Trinitiy vytvořil ze stejného souboru dat nejdelší souvislou sekvenci o délce 11 760 nukleotidů. Naopak Trinitiy vytvořil výrazně více souvislých sekvencí, konkrétně 1 329 960 kusů, než Megahit (384 096).
Zde okamžitě vyvstává otázka:
Jaká by byla navržená sekvence údajného viru SARS-CoV-2, kdyby na jedné straně neexistoval software Megahit a na druhé straně odpovídající referenční genomy (viry SARS podobné netopýrům a dříve známé viry SARS spojené s lidmi)?
Další důležitou otázkou je:
Proč je délka deklarovaného koronaviru SARS-CoV-2 pouze 29 903 nukleotidů[2], což je o 571 nukleotidů méně než nejdelší kontig, který má 30 474 nukleotidů?
Velký rozdíl mezi oběma programy, pokud jde o maximální délku kontigů, je velmi pozoruhodný. Nezdá se, že by pro algoritmy používané k vyhledávání překryvů existovala nějaká uznávaná kritéria správnosti.
To je obzvláště důležité z vědeckého hlediska, zejména s ohledem na reprodukovatelnost. Z dosavadní analýzy navíc vyplývá, zejména s ohledem na možné náhodné chyby v průběhu sekvenování nebo na detekci případně ve skutečnosti neexistujících nukleotidových sekvencí, že z elementárních logických úvah mohou být vypočtené sekvence nebo deklarované virové genomy pouze modely. Proto není přípustné hovořit o skutečném virovém genomu nebo skutečné virové sekvenci. Pojmenování této důležité skutečnosti a možných postupů pro ověřování modelů se v odborné literatuře nevyskytuje nebo jen okrajově.
V článku “Choice of assemblers has a critical impact on de novo assembly of SARS-CoV-2 genome and characterising variants” [5] autoři ukazují, že při rekonstrukci genomu SARS-CoV-2 z několika set vzorků hraje výběr assembleru významnou roli. Dále ukázali, že nejméně 9 % variant mezi Megahitem a metaSPAdesem (jiným assemblerem) je jedinečných pro metody assembleru. Vědci dospěli k závěru, že jejich analýzy ukazují, jak zásadní roli hrají metody sestavování genomů SARS-CoV-2 z krátkých čtení při charakterizaci tzv. variant.
Na toto zjištění proto navazuje mimo jiné následující otázka:
Co jsou vlastně virové varianty nebo virové mutace? Biologická realita, nebo bioinformatický artefakt?
Souhrnně lze konstatovat, že výsledky montáže vykazují velké rozdíly. V této publikaci je uvedeno, že nejdelší souvislá sekvence vypočtená pomocí Megahitu (30 474 nt) zahrnuje téměř celý virový genom. Na základě této sekvence byly navrženy primery PCR. Doplňková tabulka 8. “PCR primery použité v této studii”, [Tabulka 8] uvádí 52 primerů pro amplifikaci genomu. Ty rovnoměrně pokrývají celý deklarovaný virový genom, jak ukazuje následující graf.
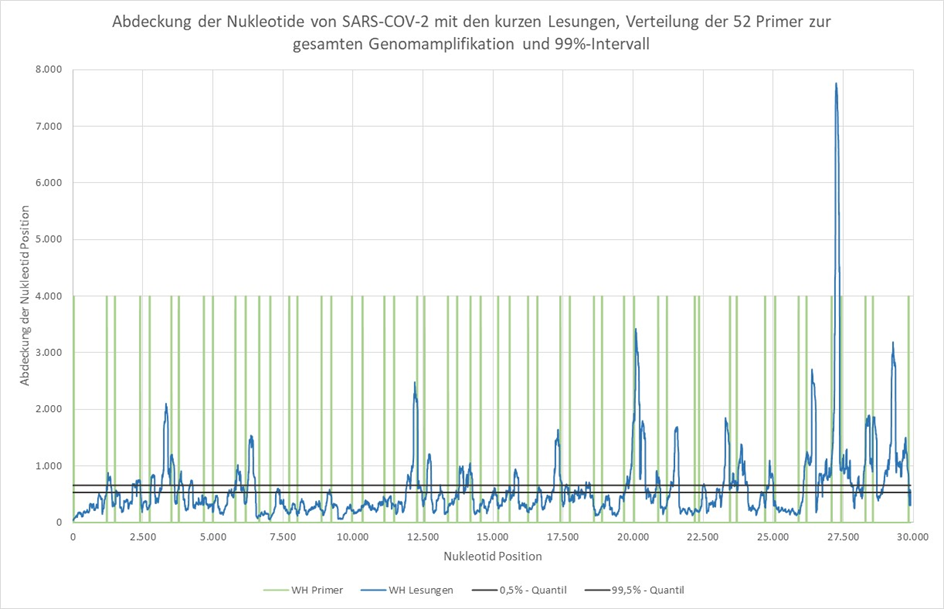
Obrázek 1: Pokrytí nukleotidů SARS-CoV-2 krátkými čteními, rozložení 52 primerů pro amplifikaci celého genomu a 99% interval. Pokrytí nukleotidů bylo stanoveno pomocí nástrojů Bowtie2 [6] a Samtools [7].
Z této publikace není zřejmé, jak přesně byla provedena konfirmace PCR nebo určení genomových parametrů. Obrázek 1 však ukazuje obzvláště vysoké pokrytí nukleotidů deklarovaného virového genomu v blízkosti uvedených primerů. Nápadná je také silně se měnící hloubka pokrytí jednotlivých nukleotidových pozic.
Za předpokladu, že se všech 29 903 nukleotidových pozic vyskytuje se stejnou pravděpodobností ve čteních spojených se SARS-CoV-2, mělo by pokrytí pro každou nukleotidovou pozici ležet mezi dvěma vodorovnými šedými čarami s pravděpodobností 99 %.
To neplatí pro přibližně 90 % nukleotidových pozic (viz modré čáry), což je pozoruhodné. A priori by se dalo očekávat, že pokud je ve vzorku přítomno dostatečné množství virové RNA a je přečteno dostatečné množství sekvenčních fragmentů, je dosaženo homogenního pokrytí nukleotidů ve virovém genomu.
Dále vyvstává otázka, proč je pro objasnění definice genomu nutný druhý, specifický krok konfirmace a stanovení na základě primerů. To vede k předpokladu, že tento krok vede ke zkreslení, které je v grafu jasně patrné.
Navíc by mohl vzniknout dojem, že použití mnoha primerů PCR specifických pro deklarovaný virový genom a amplifikace s nevědecky vysokým počtem 35 cyklů vede k “vazebným sekvencím”, které ve skutečnosti neexistují.
Tento předpoklad je podpořen sekvenováním celé RNA komerční, nikoli “infikované” kultury lidských buněk. Potřebná amplifikace fragmentů cDNA byla provedena polymerázovou řetězovou reakcí (PCR) se 14 cykly a výhradně s použitím náhodných hexamerů. To znamená krátké primery s 6 náhodnými a nespecifickými nukleotidy, což by mělo vést k relativně homogennímu pokrytí vypočtených kontigů přidruženými sekvenčními čteními.
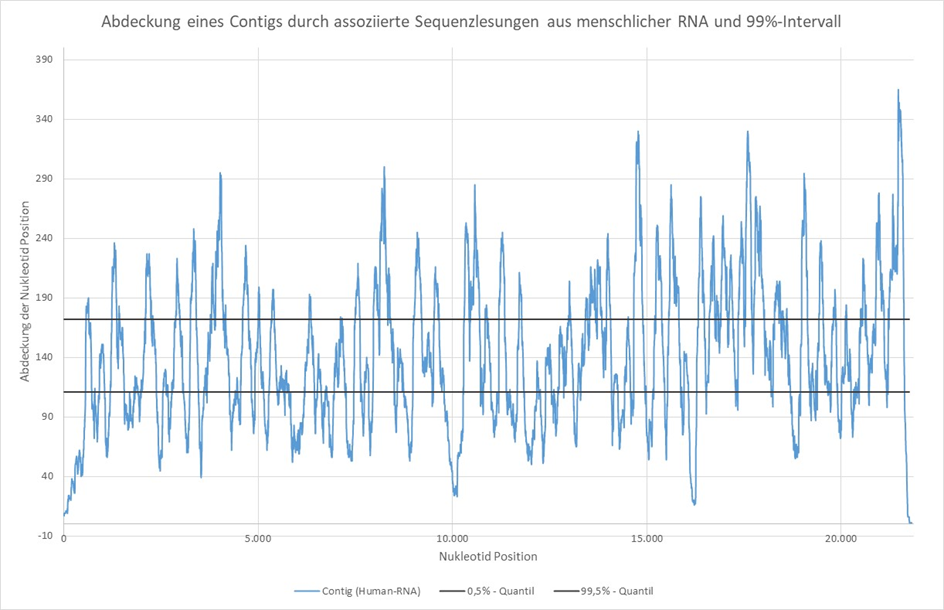
Obrázek 2: Pokrytí kontigu (21 814 nt) přidruženými sekvenčními čteními z lidské RNA a 99% intervalem. Pokrytí nukleotidů bylo stanoveno pomocí nástrojů Bowtie2 [6] a Samtools [7].
Na rozdíl od obrázku 1 ukazuje obrázek 2 mnohem homogennější pokrytí 21 814 nukleotidových pozic, i když pokrytí mnoha nukleotidových pozic leží také mimo interval 99 %. Odchylky jsou zde však s větší pravděpodobností nezkreslené pod a nad 99% intervalem.
Kontig o délce 21 814 nt, který je uveden na obrázku 2, byl vypočítán z RNA komerční lidské a “neinfikované” buněčné kultury pomocí assembleru Megahit. Porovnání pomocí Blastn s celou nukleotidovou databází ukázalo vysokou shodu (99,91 %) téměř v celé délce kontigu se sekvencí mRNA: Homo sapiens spectrin repeat containing nuclear envelope protein 2 (SYNE2), varianta transkriptu 5, mRNA, Accession: NM_182914.
V související publikaci s názvem “A novel SYNE2 mutation identified by whole exome sequencing in a Korean family with Emery-Dreifuss muscular dystrophy” [8].
v části Rusultate je uvedeno
[… nová de novo patogenní heterozygotní missense mutace (NM_182914.2: c.4858G > A; p.Ala1620Thr) genu SYNE2, která dosud nebyla popsána, byla identifikována pomocí sekvenování celého exomu u probanda a pomocí Sangerova sekvenování u jeho syna. …].
Zajímavý postřeh sám o sobě!
Nezávisle na tomto pozorování lze konstatovat, že zmíněnou sekvenci mRNA s identifikátorem NM_182914.2 bylo možné sestavit pomocí čtení fragmentů RNA z “neinfikované” buněčné kultury lidského původu, které byly před vlastním sekvenováním amplifikovány pomocí náhodných, a tedy nespecifických hexamerů pouze 14 cykly.
Potvrzení nebo stanovení pomocí PCR s použitím specifických primerů nebo použití jakýchkoli referenčních sekvencí nebylo nutné. Proto vyvstávají následující přirozené otázky.
- Proč nejdelší kontig již neobsahuje celou sekvenci virového genomu SARS-CoV-2?
- Proč je pro konečné určení sekvence zapotřebí velké množství specifických primerů a referenčních sekvencí?
- Proč je nejdelší kontig (30 474 nt) delší než deklarovaná virová sekvence SARS-CoV-2 (29 903 nt)?
Především to odporuje jedoduché logice. Pokud již bylo nalezeno 30 474 souvislých nukleotidů, měla by “pravá” sekvence genomu jistě obsahovat alespoň 30 474 nukleotidů.
V opačném případě by některé z překryvů identifikovaných systémem Megahit byly prohlášeny za nepravdivé. Ve vědecké literatuře pro to nejsou doloženy žádné normy. V následujícím textu budou představeny jednoduché kontrolní experimenty, které vyžadují pouze základní znalost operačního systému Linux, určitou míru obeznámenosti s používanými bioinformatickými počítačovými programy a publikovanými sekvenčními daty.

Výsledek: Sekvence lze stáhnout ve formátu FASTQ pomocí příkazu “fastq-dump” balíčku sratoolkit [9] a čtení PairedEnd lze pohodlně uložit do dvou textových souborů. Každý z těchto dvou textových souborů obsahuje 28 282 964 krátkých sekvencí o průměrné délce přibližně 142 bp.
Celkem tedy oba soubory obsahují 56 565 928 krátkých sekvencí. Je však nápadné, že značný počet sekvencí se skládá z N, tj. neznámých nukleotidů. [13] Mohlo by se jednat o chybná čtení nebo o následně přepsaná čtení, například čtení lidské sekvence. Z hlediska vědecké reprodukovatelnosti je třeba tento postup považovat za kritický.

Obrázek 3: 3 příklady sekvencí z 56 565 928 čtení. Čtení s identifikací @2 se skládá pouze z neznámých nukleotidů “N” a nejhoršího skóre kvality “!”.

Zdroje: [10] [11] [12]
Komentáře: Především je třeba zmínit, že software Megahit ve verzi v.1.1.3 není spustitelný na zde používaných počítačích. Proto byla použita aktuální verze v.1.2.9.
Software Trinity není spustitelný se soubory FASTQ. Proto byl program Trinity použit s nastavením – SeqType fa (FASTA). Na rozdíl od souborů FASTQ obsahují soubory FASTA pouze sekvence bez informací o kvalitě jednotlivých bází. Tyto okolnosti je třeba vzít v úvahu při analýze výsledků.
Výsledek: V následující tabulce je uveden souhrn výsledků.
*Sestavení bez použití softwaru fastp s publikovanými sekvencemi vede k celkovému počtu kontigů 29 463 a nejdelšímu kontigu 29 802 bp.

Tabulka 2: Výsledky sestavení 2. kontrolního pokusu.
Druhý kontrolní experiment ukázal nečekaný výsledek. Jak celkový počet kontigů, tak délky maximálních souvislých sekvencí se výrazně liší od publikovaných výsledků.
Pozoruhodné je také zjištění, že pomocí Trinitiy lze vypočítat delší souvislou sekvenci, než je uvedeno v publikaci [1]. Nejdelší kontigy se navíc téměř zcela shodují s publikovaným genomem “MN908947”, jak ukazuje dotaz Blastn se standardní nukleotidovou databází.
Dva nejdelší kontigy tedy v žádném případě nelze reprodukovat!
Zveřejněná sekvenční data proto nemohou být původní, nezpracovaná sekvenční data. Je pozoruhodné, že celkový počet poskytnutých sekvencí (56 565 928) odpovídá informacím v posuzované publikaci.
Jak bylo popsáno výše, byly vyřazeny sekvence RNA lidského původu. K tomuto účelu byl použit lidský referenční genom (human release 32, GRCh38.p13). Wikipedie [4] uvádí
[… Referenční lidský genom pochází od třinácti anonymních dobrovolníků z Buffala ve státě New York. Dárci byli získáváni prostřednictvím inzerátu v novinách The Buffalo News v neděli 23. března 1997. …].
Do jaké míry je referenční genom specifický pro člověka a tedy vhodný pro spolehlivé rozpoznání sekvencí RNA lidského původu? Tuto otázku může objasnit další kontrolní experiment, který přirozeně navazuje na kontrolní experiment 2.

Výsledek: Následující tabulka zobrazuje vybrané shody dotazu. Nejdelší kontig vypočtený pomocí Megahit (k141_11881) vykazuje dokonalou shodu s publikovanou sekvencí pro SARS-CoV-2 v délce 29 801 nukleotidů. Dva kontigy (k141_18050 a k141_13168) vykazují vysokou shodu s lidskými sekvencemi v délce do přibližně 6 000 nukleotidů.

Tabulka 3: Vybrané shody dotazu z porovnání 25 nejdelších kontigů z kontrolního experimentu 2 a databáze nukleotidů NCBI.
Podle popisu předmětu se jedná o ribozomální a messengerovou RNA (rRNA a mRNA). To by znamenalo, že z původních sekvencí RNA nebyly odstraněny všechny lidské sekvence vcelku, jak tvrdili čínští vědci. Nakonec je třeba poznamenat, že doplňkové tabulky 1. a 2. ke zde posuzované publikaci ukazují, že dva nejdelší kontigy (Megahit: 30 474 nt a Trinity: 11 760 nt) mají každý vysokou nukleotidovou shodu s netopýřím koronavirem SL-CoVZC45, MG772933 89,1 % a 90,4 %. Není však uvedeno, kolik nukleotidů se celkem shoduje. Bylo by docela dobře možné, že v případě Megahitu je shoda pouze na 10 000 nukleotidech z celkových 30 474 nukleotidů při 89,1 %. To by znamenalo, že 20 474 nukleotidů by nemělo významnou shodu s netopýřím koronavirem SL-CoVZC45.
Poznámka: Kontrolní experimenty
Jak již bylo uvedeno, v posuzované publikaci nebyly doloženy žádné kontrolní pokusy. Proto je třeba předpokládat, že nebyly provedeny žádné kontrolní pokusy.
Následují další zřejmé možnosti kontroly, které by mohly potvrdit získaná zjištění.

Shrnutí a závěr
Celková RNA byla extrahována ze vzorku JEDNOHO pacienta z případu onemocnění ve Wuhanu. Podle autorů byly při tomto procesu odstraněny fragmenty lidské RNA.
- Kontrolní pokus 3 ukázal, že ribozomální nebo messengerová RNA lidského původu je s velkou pravděpodobností stále přítomna v publikovaných sekvencích tvrzeného virového genomu SARS-CoV-2 s identifikací MN908947, pokud jsou databáze správně vedeny.
- Druhý kontrolní experiment ukázal, že publikované výsledky nejsou reprodukovatelné. Zveřejněné sekvence se spíše nemusí shodovat s původními sekvencemi.
- Podle popisu v této publikaci nelze předpokládat, že by bylo možné získat téměř kompletní “virový genom” pomocí Megahitu i Trinitia. Toto pozorování je znepokojivé, protože již není možné ověřit, do jaké míry se kontig 30 474 nt sestavený pomocí Megahit shoduje s netopýřím koronavirem SL-CoVZC45 a do jaké míry se odchyluje od konečného návrhu sekvence pro SARS-CoV-2.
- Nebyly zdokumentovány žádné kontrolní pokusy. Lze tedy předpokládat, že nebyly provedeny žádné kontrolní pokusy. Uvedli jsme možné kontrolní experimenty, ale náš výčet není úplný.
- Je a priori zcela nejasné, zda je onemocnění posuzovaného pacienta “virového” původu.
- Dále bylo možné prokázat, že původ fragmentů RNA použitých pro konstrukci deklarovaného virového genomu nebyl zjištěn. Mohlo by jít jednoduše o RNA lidského původu.
- Vyvstává také otázka, zda je vůbec možné objasnit původ pomocí zde použitých metod. Stručně řečeno: jsou použité sekvence “virového” původu, nebo ne? Tato otázka nevyhnutelně vede k pojmu izolace virů ve smyslu slova “izolace”. V tomto případě nebyla nalezena (virová) částice.
Jednoduchý protokol pro nalezení možné (virové) částice je zhruba následující:
- Najděte ve vzorku pacienta (zde: tekutina z bronchoalveolární laváže) “mnoho” částic stejné morfologie. Ty by měly existovat, protože virologové přisuzují tvrzeným částicím viru SARS-CoV-2 velikost přibližně 100 nm.
- Oddělte tyto částice od všeho ostatního.
- Odeberte několik různých vzorků nalezených částic.
- Všechny vzorky sekvenujte nezávisle na sobě.
- Získejte (téměř) stejnou posloupnost ve všech případech.
Ve vědeckém smyslu bychom pak popsali částici charakterizovanou její sekvencí a morfologií. Důkaz o možné patogenitě nebo přenosnosti této částice by však zatím nebyl k dispozici.
Závěrem je tedy třeba konstatovat:
Publikace “A new coronavirus associated with human respiratory disease in China” [1] neprokázala existenci “nového viru” nebo nové “virové sekvence” nebo patogenního agens. Zejména nebyla nalezena žádná příčinná souvislost s komplexem klinických příznaků studovaného pacienta.
ODKAZY
[1] A new coronavirus associated with human respiratory disease in China | Nature
Nature volume 579, pages 265–269 (2020)
[2] Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, co – Nucleotide – NCBI (nih.gov)
[3] FASTQ format. Aug. 2021. URL: https://en.wikipedia.org/wiki/FASTQ_format
[4] Reference genome. Sep. 2021.
URL: Https://en.wikipedia.org/wiki/Reference_genome
[5] Rashedul Islam u. a. „Choice of assemblers has a critical impact on de novo assembly of SARS-CoV-2 genome and characterizing variants“. In: Briefings in Bioinformatics 22.5 (2021). DOI: 10.1093/bib/bbab102.
[6] Bowtie 2. URL: Bowtie 2: fast and sensitive read alignment (sourceforge.net)
[7] Samtools. samtools/samtools: Tools (written in C using htslib) for manipulating next-generation sequencing data. URL: GitHub – samtools/samtools: Tools (written in C using htslib) for manipulating next-generation sequencing data
[8] Sook Joung Lee u. a. „A novel SYNE2 mutation identified by whole exome sequencing in a Korean family with Emery-Dreifuss muscular dystrophy“. In: Clinica Chimica Acta 506 (2020), S. 50-54. DOI: 10.1016/j.cca.2020.03.021.
[9] Ncbi. ncbi/sra-tools: SRA Tools. URL: GitHub – ncbi/sra-tools: SRA Tools
[10] OpenGene. OpenGene/fastp: An ultra-fast all-in-one FASTQ preprocessor (QC/adapters/ trimming/_ltering/splitting/merging…) URL: GitHub – OpenGene/fastp: An ultra-fast all-in-one FASTQ preprocessor (QC/adapters/trimming/filtering/splitting/merging…)
[11] Voutcn. voutcn/megahit: Ultra-fast and me mory-efficient (meta-)genome assembler. URL: https://github.com/voutcn/megahit.
[12] Trinityrnaseq. trinityrnaseq/trinityrnaseq: Trinity RNA-Seq de novo transcriptome assembly. URL: https://github.com/trinityrnaseq/trinityrnaseq
[13] Anleitung zum Download der Sequenzen, um die geschwärzten Sequenzen (ersetzt durch “N”)
1. Download der Sequenzen via fastq-dump (Konsolenprogramm für Windows oder Linux)
fastq-dump –split-files –origfmt –gzip SRR10971381
2. Falls sie nur einen teil herunterladen möchten (Ab Reads 1 bis 100, also 100 Reads:
fastq-dump –split-files -N 1 -X 100 -Z SRR10971381 > SRR10971381.fastq
3. Hier der offizielle Link https://trace.ncbi.nlm.nih.gov/Traces/sra/?run=SRR10971381
Die ersten 100 Paired-End-Reads mit dem Befehl unter 2. erzeugt. Schon unter diesen ersten 100 sind diverse Reads “geschwärzt”

14] Kontrollexperiment Phase 1 – Mehrere Labore bestätigen die Widerlegung der Virologie durch den cytopathischen Effekt | [Telegraph] (CZ verze ZDE)
Tabulka
[Tabelle 8] 41586_2020_2008_MOESM1_ESM.pdf (springer.com)

3. dubna 2022 Zdroj Přeložil: Vladimír Bartoš Když vědci ohýbají data na maximum a ostatní je následují Před několika dny jsme na našem kanálu Corona_Fakten zveřejnili náš kontrolní experiment fáze 1 (CZ verze ZDE), ve kterém jsme diskutovali o laboratorních výsledcích takzvaného cytopatickém efektu. Získané výsledky jasně ukazují,…